Introduction
People describe DNA in many ways. Bill Gates has likened it to a computer program, though it far surpasses the complexity of any man-made software. Another depicts DNA as its own language, telling the story of life itself. Even the pages of a biology textbook may describe DNA as the blueprint for life. All these representations – a computer software, a linguistic masterpiece, or an architectural guide – come from the unwavering truth that our DNA is fundamental to our very existence. Indeed, DNA shapes our every facet, from the intricate biochemical processes within microscopic cells to the broad impacts of evolution.
The Distinctive Nature of Human DNA
In particular, human DNA is unique and remarkably complex, a testament to its fundamental role in shaping our lives. We have 23 pairs of chromosomes: 22 pairs of autosomes (chromosomes unrelated to sex determination) and one pair of sex chromosomes. Autosome number and type are consistent across all humans, but sex chromosomes vary between individuals. In biological females (XX), there are two X sex chromosomes and in males (XY) there is one X and one Y sex chromosome. To counteract the different levels of gene expression across sexes, a process called X chromosome inactivation randomly silences one of the X chromosomes in females, resulting in an active (Xa) and inactive (Xi) X chromosome.4

As a result of X chromosome inactivation, mutations in the sex chromosomes have different implications compared to mutations in autosomes. While the X chromosome only consists of 4% of the human genome, it is the origin of more than 20% of neurological disabilities, a disparity that has attracted the attention of researchers and scientists. Since biological males receive only a single copy of the X chromosome, they lack the alternative secondary copy that females have; as a result, X-linked genetic disorders have more serious symptoms in males, often having detrimental effects on their development. Females can still be impacted by these disorders, though to a lesser extent. In females, cells with a sex chromosome mutation on the inactive X chromosome have a selective disadvantage in development, resulting in a higher population of cells with the normal X chromosome active. This in turn leads to milder symptoms of X-linked disorders and a relatively healthy female individual, since the majority of the mutated X chromosomes are not expressed.4

The Neuroanatomical and Symptomatic Impacts of Fragile-X Syndrome
One of the most commonly inherited forms of autism spectrum disorder (ASD) is known as Fragile-X syndrome (FXS), which results from mutation of the Fragile-X mental retardation 1 (FMR1) gene located on the X chromosome. Approximately one in every 7,000 males and one in every 11,000 females are diagnosed with this mutation. Nearly all males diagnosed with FMRI mutation have an intellectual disability, hyperactivity, attention problems, and anxiety. Around half of this group have also been found to have ASD. In females, the symptoms are milder with around half of all diagnosed females facing serious intellectual disability and anxiety.3
The Genetic Basis of Fragile-X Syndrome
The genome is composed of DNA, a double stranded helical molecule. DNA encodes important proteins necessary to regulate or perform cellular functions through sequences of nucleotide molecules, which consist of a phosphate-sugar backbone and one of four nitrogenous bases (adenine: A, guanine: G, cytosine: C, and thymine: T).
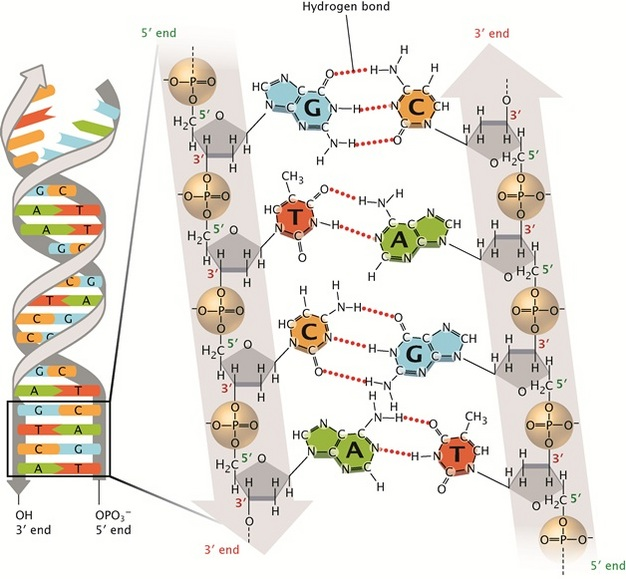
The exact DNA sequences that encode functional messenger RNA (the molecule that synthesizes the critical proteins that drive cellular functions) lie within regions of the chromosome called genes. These genes primarily consist of introns (noncoding regions) and exons (coding regions), which are later converted into RNA in a process known as transcription. During cellular processing of RNA, introns are frequently spliced out of the final RNA transcript, while exons are joined together.
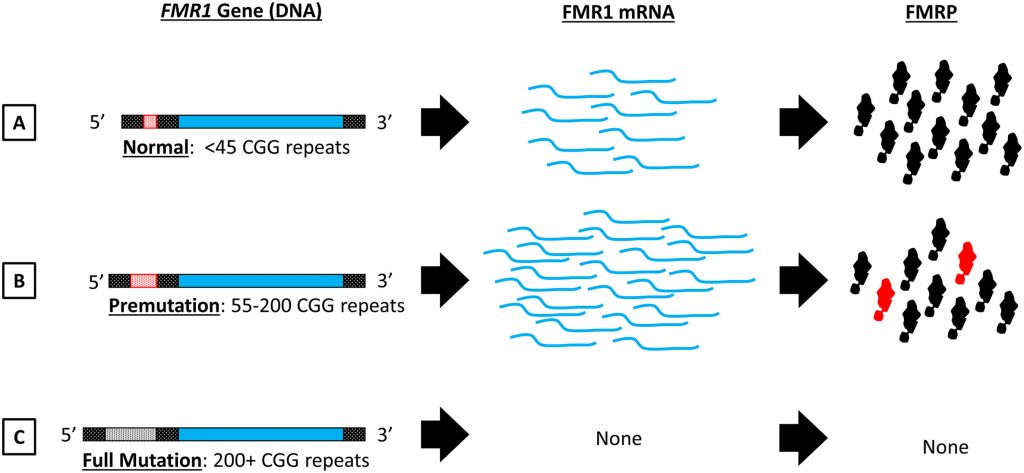
The basis for FXS involves a mutation in the FMR1 gene caused by a genetic mutation that consists of a specific expansion of CGG nucleotide repeats in the untranslated (non-protein encoding) region of the gene.1,3 The number of repeats varies across individuals but the number normally ranges on average from 15 to 45 repeats. However, in individuals with FXS, the number of CGG repeats is 200 or more. The methylation, or addition of methyl groups to DNA nucleotides, to these repeats represses the transcription of the FMR1 gene.5 This leads to little or none of the FMR protein, depending on the number of DNA repeats. This protein is an RNA binding protein that negatively regulates local protein synthesis in the dendrites of neurons, which are the outward, branchlike cellular extensions responsible for receiving electrical signal output from neighboring neurons. In individuals diagnosed with FXS, it has been observed that neurons in certain brain areas are immature, and are characterized by overgrowth in dendritic spines. These dendritic spines are protrusions on dendrites and directly receive electrical input. In FXS patients, they have been found to be abnormally long, leading scientists to believe that a mutation of the FMR1 gene ultimately disrupts dendritic spine development.3
The Potential Applications of Gene Therapy for Fragile-X Syndrome Treatment
Despite the extensive research characterizing FXS, there is currently no treatment that directly cures the disorder. Current treatments require early intervention and only alleviate the symptoms of the condition.6 However, there has been promising research utilizing gene therapies utilizing a peptide derived from an HIV protein called Tat. This peptide aids in the entry of therapeutic molecules into the cell to restore the function of the cell intracellularly.9 In particular, a modified FMRP protein called a tat-conjugated N-terminal fragment of FMRP (FMRP-N-tat) has been found to restore function in cells containing a knockout gene of FMR1 that do not endogenously have FMRP protein.8 Additionally, modified FMRP protein was found to improve the elevated activity levels of FXS mice models.7 One day, researchers hope to utilize these gene therapies to develop a noninvasive, effective treatment for individuals diagnosed with FXS.
Conclusion
DNA is more than just a blueprint or computer program, but rather holds the answers to many biological questions in its structure and function. The Fragile X Syndrome is one of the many neurodevelopmental disorders that stem from genetic mutations in DNA. Through unpacking the precise genetic and neural circuits of disease pathology using modern genetic technologies, scientists are capable of developing novel treatments to more directly target the exact molecular underpinnings of potentially devastating symptoms.
Acknowledgements
I would like to thank Professor Paul J. Hagerman and Professor Randi J. Hagerman for reviewing my article for accuracy. I would also like to thank the BSJ editorial staff for peer reviewing my article.
References
- Garber, K. B., Visootsak, J., & Warren, S. T. (2008). Fragile X syndrome. European Journal of Human Genetics : EJHG, 16(6), 666–672. https://doi.org/10.1038/ejhg.2008.61
- Grimm, N.-B., & Lee, J. T. (2022). Selective Xi Reactivation and Alternative Methods to Restore MECP2 Function in Rett Syndrome. Trends in Genetics : TIG, 38(9), 920–943. https://doi.org/10.1016/j.tig.2022.01.007
- Salcedo-Arellano, M. J., Dufour, B., McLennan, Y., Martinez-Cerdeno, V., & Hagerman, R. (2020). Fragile X syndrome and associated disorders: Clinical aspects and pathology. Neurobiology of Disease, 136, 104740. https://doi.org/10.1016/j.nbd.2020.104740
- Sun, Z., Fan, J., & Wang, Y. (2022). X-Chromosome Inactivation and Related Diseases. Genetics Research, 2022, 1391807. https://doi.org/10.1155/2022/1391807
- Sutcliffe, J. S., Nelson, D. L., Zhang, F., Pieretti, M., Caskey, C. T., Saxe, D., & Warren, S. T. (1992). DNA methylation represses FMR-1 transcription in fragile X syndrome. Human Molecular Genetics, 1(6), 397–400. https://doi.org/10.1093/hmg/1.6.397
- What are the treatments for Fragile X syndrome? | NICHD – Eunice Kennedy Shriver National Institute of Child Health and Human Development. (2021, August 5). https://www.nichd.nih.gov/health/topics/fragilex/conditioninfo/treatments
- Zhan, X., Asmara, H., Cheng, N., Sahu, G., Sanchez, E., Zhang, F.-X., Zamponi, G. W., Rho, J. M., & Turner, R. W. (2020). FMRP(1-297)-tat restores ion channel and synaptic function in a model of Fragile X syndrome. Nature Communications, 11(1), 2755. https://doi.org/10.1038/s41467-020-16250-4
- Zhan, X., Asmara, H., Pfaffinger, P., & Turner, R. W. (2024). Calcium-dependent regulation of neuronal excitability is rescued in Fragile X Syndrome by a tat-conjugated N-terminal fragment of FMRP. Journal of Neuroscience. https://doi.org/10.1523/JNEUROSCI.0136-24.2024
- Zhang, P., Lock, L. L., Cheetham, A. G., & Cui, H. (2014). Enhanced Cellular Entry and Efficacy of Tat Conjugates by Rational Design of the Auxiliary Segment. Molecular Pharmaceutics, 11(3), 964–973. https://doi.org/10.1021/mp400619v
Image References
Banner Image: A close up of a blue and purple structure photo – Free 3d Image on Unsplash. (n.d.). Retrieved March 18, 2024, from https://unsplash.com/photos/a-close-up-of-a-blue-and-purple-structure-8o_LkMpo8ug
Figure 1: Calico cat. (2024). In Wikipedia. https://en.wikipedia.org/w/index.php?title=Calico_cat&oldid=1208404448
Figure 2: X-inactivation. (2024). In Wikipedia. https://en.wikipedia.org/w/index.php?title=X-inactivation&oldid=1209097412
Figure 3: Discovery of DNA Double Helix: Watson and Crick | Learn Science at Scitable. (n.d.-a). Retrieved March 2, 2024, from http://www.nature.com/scitable/topicpage/discovery-of-dna-structure-and-function-watson-397