Introduction
By the time you finish reading this sentence, your body will have produced 38 million new cells.1 Within these cells exist billions of mutations, or structural alterations of your DNA. If that sounds scary, you needn’t worry — the body follows its genetic code as closely as an orchestra does its sheet music. But while the body consistently maintains harmony, it’s conducting a concert while simultaneously tuning its instruments. Mutations are a way to add richness to individual genetic expression, to the extent that each of us was born with seventy de novo mutations not observed in either parent. Most mutations are never activated or inherited, while others can present as benign (like heterochromia) or even beneficial (like lactose tolerance), making them a key component of our evolution as a species.
When mutations are harmful, they are usually eliminated quickly—fractions of a fraction of a second quickly. However, sometimes mutations can evade the body’s natural surveillance system; because mutations generate within the body’s cells rather than invade the body like infectious diseases, affected cells can avoid detection until they reproduce, accumulating mutations to the point they spiral out of control and become tumors. Mutations with “detectable effects (i.e., activating, inactivating, inhibitory, and non-inhibitory)” are known as driver mutations because they drive the generation and/or the spread of cancer.2 Mutations with “undetectable” or neutral effects are considered passenger mutations since they are just along for the ride. The ratio of driver-to-passenger mutations is what differentiates malignant from benign tumors.
It is difficult for scientists to find solutions for harmful mutations, as while mutations are inevitable and not entirely random, they’re notoriously unpredictable. However, recent advancements in single-cell RNA sequencing technology (scRNA-seq) have allowed a clearer peek into the enigmatic machinations of mutations, which might eventually allow more efficient and effective cancer treatment.
Background
Single-cell RNA sequencing examines individual cells’ RNA. While DNA contains the code for our bodies to perform all its operations, RNA is what helps carry out these instructions by serving as the blueprint for specialized cell types; while every cell in our bodies has the same DNA, each type bears different mRNA molecules. As such, sequencing RNA allows researchers to understand how a cell replicates and behaves. Therefore, using scRNA-seq on tumor cells would allow researchers to understand how those abnormal cells function.
ScRNA-seq copies cell RNA into complementary DNA (cDNA). This complementary DNA (cDNA) is then amplified, or reproduced, many times to provide as much insight into a genome, or the complete set of genes in an organism, as possible.3 From there, researchers can analyze and map the sequence of nucleotides (DNA building blocks) in the cDNA into a “network of immune checkpoints,” within samples. The collected readout of all a cell’s RNA molecules is known as a transcriptome. Transcriptomes help to understand what triggers certain genetic expressions by recording what checkpoints are activated during intercellular and environmental interactions.3
Transcriptomes allow a tumor’s complex microenvironment of varied cells to be more easily navigated. In this sense, a transcriptome is as crucial to cancer researchers as an atlas is to a traveler, allowing noteworthy cell activity to be readily detected, identified, and addressed. In addition, it allows researchers to quickly differentiate cell types within the microenvironment of a tumor.
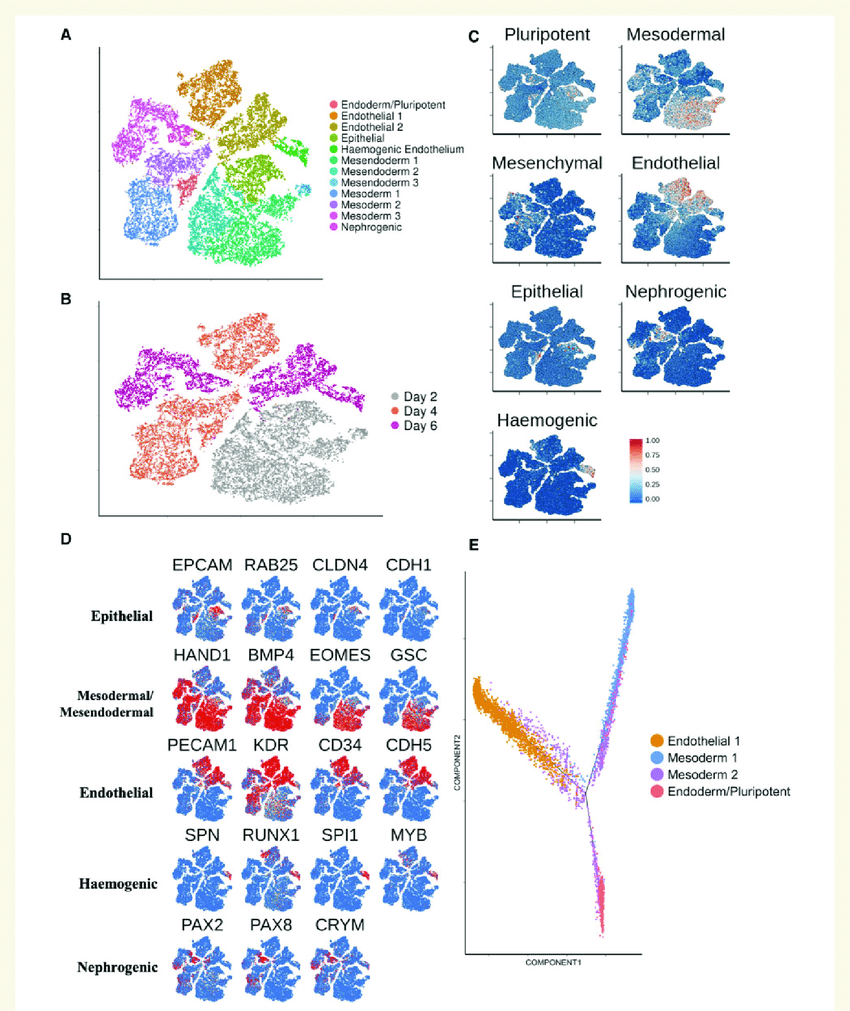
This ultimately contributes towards our understanding of the precise details on how different cells mutate and become metastatic (spreading the cancer to other parts of the body). This activity can be cataloged, refining transcriptomes to record even more immune checkpoints and increase sensitivity in the event a cell mutates. This allows quicker and more thorough responses compared to previous generations of bulk-RNA sequencing technology, which could only record a few dozen markers at a time without identifying which cell type they came from4. Because cancer treatment extends over months or years, the chance of tumors developing drug resistance increases over time. Understanding this tumor adaptation is critical to developing new therapies that outpace them. ScRNA-seq removes much of the room for error with its specificity, which may allow cancer treatments to be tailored to single individuals and cell types.
Finding Footing
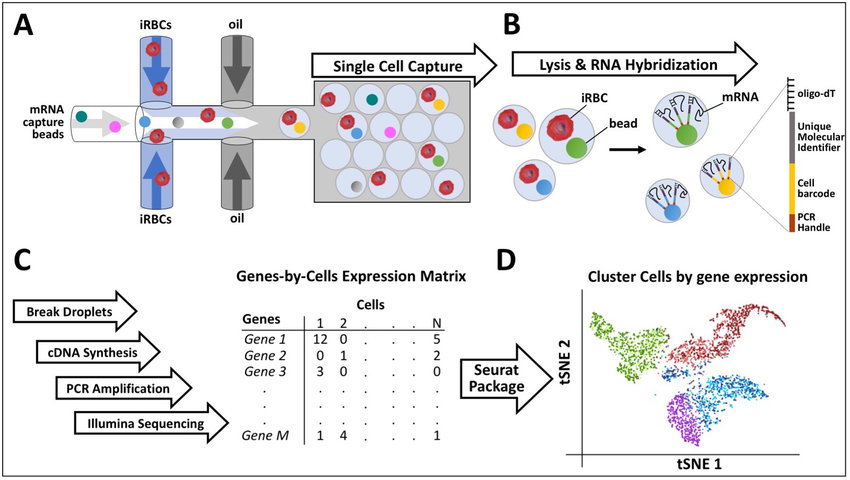
Because scRNA-seq is a rapidly expanding field of study, techniques for its procedure are constantly evolving. Still, the most common strategies for its procedure are “droplet-based” and “microwell” sequencing, according to a recent study by the University of Rostock, Germany5.
Droplet-based sequencing involves encasing a cell alongside a specialized bead in a drop of microfluid, which is dozens or even hundreds of times less volume than one molecule of H2O. Beads bind to cells by being emulsified with a kind of oil.
What makes DropSeq notable is the precision of its transcription. As explained by Dr. Eric Chow, Director of the Center for Advanced Technology at the University of California, San Francisco, DropSeq techniques perform transcription within the drops themselves, which is faster and more fine-tuned than scanning a cell from the outside-in.4 As each bead bears a unique barcode that copies onto cDNA, this method also ensures no readouts would be confused with one another.
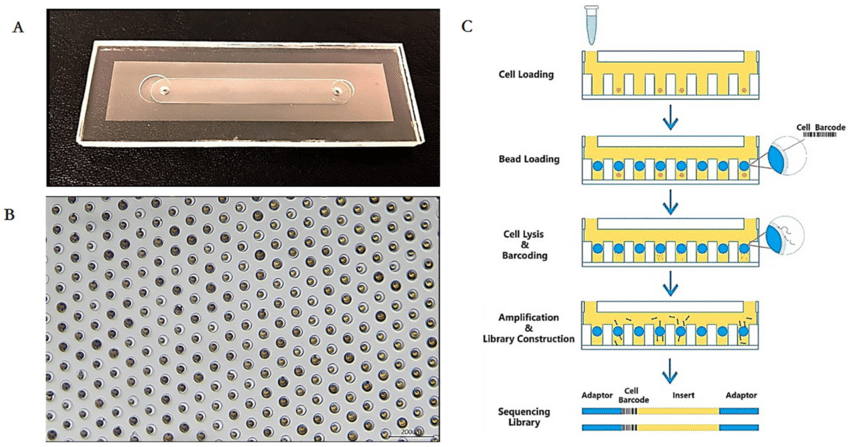
Microwell sequencing involves organizing cells into microscopic wells on a chip. A single chip can hold tens of thousands or even hundreds of thousands of wells at once, which all contain a bead that captures its paired cell’s RNA, similar to DropSeq.5 Where DropSeq returns high throughput through its process of cDNA amplification, microwell sequencing is advantageous for its “ability to visually inspect captured cells, allowing for identification of wells containing damaged or no cells and/or providing additional morphological information”5. Because the cells are neatly arranged in wells, they can be uniformly scanned using lasers, whereas DropSeq techniques must rely on chance to emulsify beads and cells.
Applications
ScRNA-seq is advantageous for its high specificity and ability to differentiate between cell types, creating extremely detailed transcriptomes that catalog both genetic expression and cell-to-cell communication. This allows scRNA-seq to pair well with immunotherapy, tailoring cancer remedies to specific cells in tumor microenvironments. In this sense, creating treatment based on scRNA-seq is like using a sniper to take out tumors, whereas traditional forms of chemotherapy and radiotherapy raze body parts experiencing metastasis, killing native cells and tumor cells indiscriminately.
Oncologists have also been able to refine their classifications of subtypes of cancer through transcriptomes, such as in a 2020 study that discovered a subtype of lung adenocarcinoma, which affects the glands that produce tissue linings, mucus, and other fluids.6 This same study found that genes can be reprogrammed using certain drugs known as kinase inhibitors, which interfere with cells’ abilities to communicate and replicate. To understand this drug’s function, imagine searching for a specific phrase in a long screed of a document. You could skim the whole thing in your search, or you could use Ctrl+F and look up keywords and edit them. Kinase inhibitors are similar to this latter option.
The benefits of transcriptomes in medicine have been furthered by a 2022 study that discovered the gene S100A4 was “implicated in several chronic inflammatory diseases including rheumatoid arthritis, asthma, and allergies…poor survival in glioma as well as in breast, bladder, head and neck, and pancreatic cancers, and…necessary for breast cancer metastasis,” making it a key player in the development of disease.6 S100A4 is just one gene, yet it exerts so much control over the body. Through scRNA-seq, geneticists understood how S100A4 worked and figured out ways to regulate, limit, and even silence its expression by altering its transcription in the genome. As a result of targeting S100A4 in an immunotherapy regimen on mice, “[tumor-bearing] mice [were found to] live significantly longer than B6 wild-type host mice,” with no observations of “spontaneous tumors in [S100A4 colonies] in over a decade,” which bodes well for its applicability to human patients.6
We can take this beyond reprogramming genes to the ability to predict mutations themselves; there are currently thirty-three scRNA-seq algorithms capable of predicting driver mutations.2 Using machine learning to cross-reference transcriptome readouts with gene activity in real-time, these algorithms learn to distinguish between driver mutations and passenger mutations through each step of their activation, accumulation, and reproduction — ultimately, they become able to recognize which cells are liable to aggressive mutations based on their protein activity. As proteins are critical to the organization and maintenance of a cell, their decline is the best clue to the overall health of a cell. Programming these algorithms based on transcriptomes allows further insight into how cell activity increases or decreases the chances of developing driver mutations, allowing cancer to be detected earlier and thus treated with less aggressive regimens.
The Next Step
Deaths from cancer push half a million deaths annually, making it the second leading cause of death in the United States behind heart disease7.
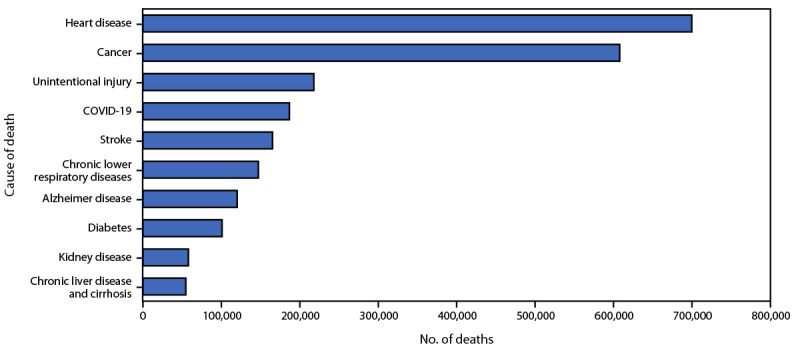
Curing cancer is a challenge because the combination of responsible mutations is different between regions of one tumor, let alone between people. However, cancer treatments continue to refine each decade, cutting down time spent in the clinic while extending patients’ lifespans. ScRNA-seq contributes not only to advancements in cancer treatments but also the potential for earlier detection, reversal. In all, advancements like these reduce the terror of cancer by making it more manageable and less painful to treat.
Acknowledgements
I’d like to thank Claudia Cattoglio, a research specialist for the Tjian + Darzacq Group at UC Berkeley, for lending her expertise on molecular dynamics in reviewing this piece.
References
[1] Fischetti, M, Christiansen, J. (2021, April 1). Our Bodies Replace Billions of Cells Every Day. Blood and the gut dominate cell turnover. Scientific American Magazine. https://www.scientificamerican.com/article/our-bodies-replace-billions-of-cells-every-day/
[2] Chen, H., Li, J., Wang, Y. et al. Comprehensive assessment of computational algorithms in predicting cancer driver mutations. Genome Biol 21, 43 (2020). https://doi.org/10.1186/s13059-020-01954-z
[3] Zhang, Y., Wang, D., Peng, M. et al. Single‐cell RNA sequencing in cancer research. J Exp Clin Cancer Res 40, 81 (2021). https://doi.org/10.1186/s13046-021-01874-1
[4] iBiology Techniques. (2020, August 28). Single Cell Sequencing – Eric Chow (UCSF) [Video]. https://www.youtube.com/watch?v=k9VFNLLQP8c
[5] Wolfien M, David R, Galow AM. Single-Cell RNA Sequencing Procedures and Data Analysis. In: Helder I. N, editor. Bioinformatics [Internet]. Brisbane (AU): Exon Publications; 2021 Mar 20. Chapter 2. Available from: https://www.ncbi.nlm.nih.gov/books/NBK569559/ doi: 10.36255/exonpublications.bioinformatics.2021.ch2
[6] Kim, N., Kim, H. K., Lee, K., Hong, Y., Cho, J. H., Choi, J. W., Lee, J., Suh, Y., Ku, B. M., Eum, H. H., Choi, S., Choi, Y., Joung, J., Park, W., Jung, H. A., Sun, J., Lee, S., Ahn, J. S., Park, K., . . . Lee, H. (2020). Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nature Communications, 11(1), 1-15. https://doi.org/10.1038/s41467-020-16164-1
[7] Ahmad, F. B., Cisewski, J. A., Xu, J., & Anderson, R. N. (2023). Provisional Mortality Data — United States, 2022. Morbidity and Mortality Weekly Report, 72(18), 488-492. https://doi.org/10.15585/mmwr.mm7218a3
[8] You, G., Zheng, Z., Huang, Y., Liu, G., Luo, W., Huang, J., Zhuo, L., Tang, B., Liu, S., & Lin, C. (2023). ScRNA-seq and proteomics reveal the distinction of M2-like macrophages between primary and recurrent malignant glioma and its critical role in the recurrence. CNS Neuroscience & Therapeutics, 29(11), 3391-3405. https://doi.org/10.1111/cns.14269
Image References
Figure 1: McCracken, Ian & Taylor, Richard & Kok, Fatma & De la Cuesta, Fernando & Dobie, Ross & Henderson, Beth & Mountford, Joanne & Caudrillier, Axelle & Henderson, Neil & Ponting, Chris & Baker, Andrew. (2019). Transcriptional dynamics of pluripotent stem cell-derived endothelial cell differentiation revealed by single-cell RNA sequencing. European Heart Journal. 41. 10.1093/eurheartj/ehz351.
Figure 2: Poran, Asaf & Nötzel, Christopher & Aly, Omar & Mencia Trinchant, Nuria & Harris, Chantal & Guzman, Monica & Hassane, Duane & Elemento, Olivier & Kafsack, Björn. (2017). Single-cell RNA sequencing reveals a signature of sexual commitment in malaria parasites. Nature. 551. 10.1038/nature24280.
Figure 3: Li, Minli & Liu, Hongde & Guo, Yunxia & Chen, Fang & zi, Xiaoyuan & Fan, Rong & 李华梅, Ksc & Cai, Yiran & He, Chunpeng & Lu, Zuhong. (2020). Single symbiotic cell transcriptome sequencing of coral. Genomics. 112. 10.1016/j.ygeno.2020.10.019.
Figure 4: Ahmad, F. B., Cisewski, J. A., Xu, J., & Anderson, R. N. (2023). Provisional Mortality Data — United States, 2022. Morbidity and Mortality Weekly Report, 72(18), 488-492. https://doi.org/10.15585/mmwr.mm7218a3